Introduction to SJD (Structured Joint Decomposition)
Huan Chen
Jinrui Liu
Shreyash Sonthalia
Guangyan Li
Carlo Colantuoni
02 October, 2022
Source:vignettes/StructralJointDecomposition.Rmd
StructralJointDecomposition.RmdWe are using a list of neurogenesis datasets of both human and mouse model from different studies to demonstrate the use of SJD package. Here we have a list of datasets NeuroGenesis4, and one dataframe NeuroGenesis.info containing meta informationtot helps visualize the results. Users are suggested to make the dataframe including information prior to visualization.
Install and load SJD package
To install this package in R, run the following commands:
library(devtools)
install_github("CHuanSite/SJD")Jointly decompose 4 NeuroGenesis genomics data with SJD
package
Load data
The whole structured NeuroGenesis4 and its meta information NeuroGenesis.info are deposited to googledrive and the downsized subsets are built in the SJD package. Please follow the below commands to download the real data and info For users who wants to save time by using the subset data, please skip downloading from google drive and follow the commands below.
We download the full data sets from the google drive, and load it
into R
install.packages("googledrive", repos = "http://cran.us.r-project.org")
#> Installing package into '/private/var/folders/hn/15_w90p96zn9px58h76x8sfr0000gn/T/Rtmpnf4wal/temp_libpath14ecf600e2706'
#> (as 'lib' is unspecified)
#>
#> The downloaded binary packages are in
#> /var/folders/hn/15_w90p96zn9px58h76x8sfr0000gn/T//RtmpbEWihf/downloaded_packages
library(googledrive)
## Download NeuroGenesis4
drive_download(file = "https://drive.google.com/u/0/uc?export=download&id=1jf4f_dxps47yT89m8Tkpviyci_XoOtGk", path = "./NeuroGenesis4.RData", overwrite = TRUE)
#> ! Using an auto-discovered, cached token.
#> To suppress this message, modify your code or options to clearly consent to
#> the use of a cached token.
#> See gargle's "Non-interactive auth" vignette for more details:
#> <https://gargle.r-lib.org/articles/non-interactive-auth.html>
#> ℹ The googledrive package is using a cached token for hzchenhuan@gmail.com.
#> Auto-refreshing stale OAuth token.
#> File downloaded:
#> • NeuroGenesis4_full_data.RData <id: 1jf4f_dxps47yT89m8Tkpviyci_XoOtGk>
#> Saved locally as:
#> • ./NeuroGenesis4.RData
## Download NeruGenesis4.info
drive_download(file = "https://drive.google.com/file/d/1aGYAxZDYJKOLLO1LmFk9vPYUmtmS1TjZ/view?usp=sharing", path = "./NeuroGenesis4.info.RData", overwrite = TRUE)
#> File downloaded:
#> • NeuroGenesis4_full_info.RData <id: 1aGYAxZDYJKOLLO1LmFk9vPYUmtmS1TjZ>
#> Saved locally as:
#> • ./NeuroGenesis4.info.RData
load(file = "./NeuroGenesis4.RData")
load(file = "./NeuroGenesis4.info.RData")This block of codes containing the small sampled data, which is faster to compute than the full scale data,
# data("NeuroGenesis4", package = "SJD")
# data("NeuroGenesis4.info", package = "SJD")
# str(NeuroGenesis4)Preprocess data
Here we input the list of datasets (expression matrices) and wrap them to be ready for SJD analysis. sjdWrap identifies the shared genes among all datasets regardless of species and output a new list of datasets with uniformed gene names
- dataset_list: expression matrices to be analyzed
- species_vetor: the species information for each input dataset
- geneType_vetor: the gene type information for each input dataset
- geneType_out: the desired gene type for all datasets
- species_out: the desired species to refer when uniform the genes for all datasets
SJDdataIN = sjdWrap(
data.list = NeuroGenesis4,
species.vector = c("human", "human", "human", "mouse"),
geneType.vector = c("symbol","ensembl","symbol","symbol"),
geneType.out = "symbol",
species.out = "human"
)
#> Using biomaRt to connect gene IDs across 4 datasets:
#> Getting biomaRt IDs for dataset 1
#> You have input 23368 genes
#> We found 22458 matches
#> 2624 of those are duplicates and only keeping the 1st of each
#> Getting biomaRt IDs for dataset 2
#> You have input 63677 genes
#> We found 54544 matches
#> 17816 of those are duplicates and only keeping the 1st of each
#> Getting biomaRt IDs for dataset 3
#> You have input 35543 genes
#> We found 25753 matches
#> 2297 of those are duplicates and only keeping the 1st of each
#> Getting biomaRt IDs for dataset 4
#> You have input 16441 genes
#> We found 12475 matches
#> 637 of those are duplicates and only keeping the 1st of each
#> constructed 4 tables of cross-species matching genes
#> we found 10694 shared genes in 4 datasets
#> new data list of 4 datasets constructedApply the decomposition method
After the transformation and alignment among the data sets, the next step is to apply the decomposition method, we give examples on different methods to illustrate the process
grp = list(
Shared.All.4 = c(1 : 4),
Shared.bulk.2 = c(1,2),
Shared.sc.2 = c(3, 4),
Hs.Meisnr.1 = c(1),
Hs.AZ.1 = c(2),
Gesch.1 = c(3),
Telley.1 = c(4))
dims = c(2, 2, 2, 2, 2, 2, 2) # must have same length as "grp"
# sep
# sepPCA.out = sepPCA(SJDdataIN, dims)
# sepICA.out = sepICA(SJDdataIN, dims)
# sepNMF.out = sepNMF(SJDdataIN, dims)
# concat
# concatPCA.out = concatPCA(SJDdataIN, grp, dims)
# concatICA.out = concatICA(SJDdataIN, grp, dims)
# concatNMF.out = concatNMF(SJDdataIN, grp, dims)
# joint
# jointPCA.out = jointPCA(SJDdataIN, grp, dims)
# jointICA.out = jointICA(SJDdataIN, grp, dims)
# jointNMF.out = jointNMF(SJDdataIN, grp, dims)
# sequential
twoStageLCA.out = twoStageLCA(dataset = SJDdataIN, group = grp, comp_num = dims)
# twoStageiLCA.out = twoStageiLCA(dataset = SJDdataIN, group = grp, comp_num = dims)Plot
library(ggplot2)
library(gridExtra)
#>
#> Attaching package: 'gridExtra'
#> The following object is masked from 'package:Biobase':
#>
#> combine
#> The following object is masked from 'package:BiocGenerics':
#>
#> combinePlot each dim in each dataset individually
Here we need to call the SJDScorePlotter function to
generate the SJDScorePlotter.obj, a list of ggplot objects
that can be used
SampleMetaNamesTable = data.frame(
row.names = names(NeuroGenesis4),
Type = c('Yaxis','Yaxis','2Dscatter','2Dscatter'),
XaxisColumn = c("X","DAYx","tSNE_1","tsne1:ch1"),
YaxisColumn = c("PJDscores","PJDscores","tSNE_2","tsne2:ch1"),
COLaxisColumn = c("color","colorBYlabelsX","PJDscores","PJDscores"),
PCHColumn = c("","","",""),
cexx=c(1,1,2,2)
)
SJDScorePlotter.obj = SJDScorePlotter(
SJDalg = "twoStageLCA",
scores = twoStageLCA.out$score_list,
lbb = "NeuroGenesis4.p2",
info = NeuroGenesis4.info,
SampleMetaNamesTable = SampleMetaNamesTable
)
#> [1] "**********************************************************"
#> [1] "plotting scores for twoStageLCA anaysis."
#> [1] "*******************************"
#> [1] "dataset: Meissner.inVitro.bulk.Hs"
#> [1] "grouping: Shared.All.4"
#> [1] 1
#> [1] 2
#> [1] "grouping: Shared.bulk.2"
#> [1] 1
#> [1] 2
#> [1] "grouping: Shared.sc.2"
#> [1] "No SJD scores for grouping : Shared.sc.2, moving to the next SJD output for Meissner.inVitro.bulk.Hs"
#> [1] "grouping: Hs.Meisnr.1"
#> [1] 1
#> [1] 2
#> [1] "grouping: Hs.AZ.1"
#> [1] "No SJD scores for grouping : Hs.AZ.1, moving to the next SJD output for Meissner.inVitro.bulk.Hs"
#> [1] "grouping: Gesch.1"
#> [1] "No SJD scores for grouping : Gesch.1, moving to the next SJD output for Meissner.inVitro.bulk.Hs"
#> [1] "grouping: Telley.1"
#> [1] "No SJD scores for grouping : Telley.1, moving to the next SJD output for Meissner.inVitro.bulk.Hs"
#> [1] "*******************************"
#> [1] "dataset: LIBD.AZ.inVitro.bulk.Hs"
#> [1] "grouping: Shared.All.4"
#> [1] 1
#> [1] 2
#> [1] "grouping: Shared.bulk.2"
#> [1] 1
#> [1] 2
#> [1] "grouping: Shared.sc.2"
#> [1] "No SJD scores for grouping : Shared.sc.2, moving to the next SJD output for LIBD.AZ.inVitro.bulk.Hs"
#> [1] "grouping: Hs.Meisnr.1"
#> [1] "No SJD scores for grouping : Hs.Meisnr.1, moving to the next SJD output for LIBD.AZ.inVitro.bulk.Hs"
#> [1] "grouping: Hs.AZ.1"
#> [1] 1
#> [1] 2
#> [1] "grouping: Gesch.1"
#> [1] "No SJD scores for grouping : Gesch.1, moving to the next SJD output for LIBD.AZ.inVitro.bulk.Hs"
#> [1] "grouping: Telley.1"
#> [1] "No SJD scores for grouping : Telley.1, moving to the next SJD output for LIBD.AZ.inVitro.bulk.Hs"
#> [1] "*******************************"
#> [1] "dataset: Geschwind.inVivo.sc.Hs"
#> [1] "grouping: Shared.All.4"
#> [1] 1
#> [1] 2
#> [1] "grouping: Shared.bulk.2"
#> [1] "No SJD scores for grouping : Shared.bulk.2, moving to the next SJD output for Geschwind.inVivo.sc.Hs"
#> [1] "grouping: Shared.sc.2"
#> [1] 1
#> [1] 2
#> [1] "grouping: Hs.Meisnr.1"
#> [1] "No SJD scores for grouping : Hs.Meisnr.1, moving to the next SJD output for Geschwind.inVivo.sc.Hs"
#> [1] "grouping: Hs.AZ.1"
#> [1] "No SJD scores for grouping : Hs.AZ.1, moving to the next SJD output for Geschwind.inVivo.sc.Hs"
#> [1] "grouping: Gesch.1"
#> [1] 1
#> [1] 2
#> [1] "grouping: Telley.1"
#> [1] "No SJD scores for grouping : Telley.1, moving to the next SJD output for Geschwind.inVivo.sc.Hs"
#> [1] "*******************************"
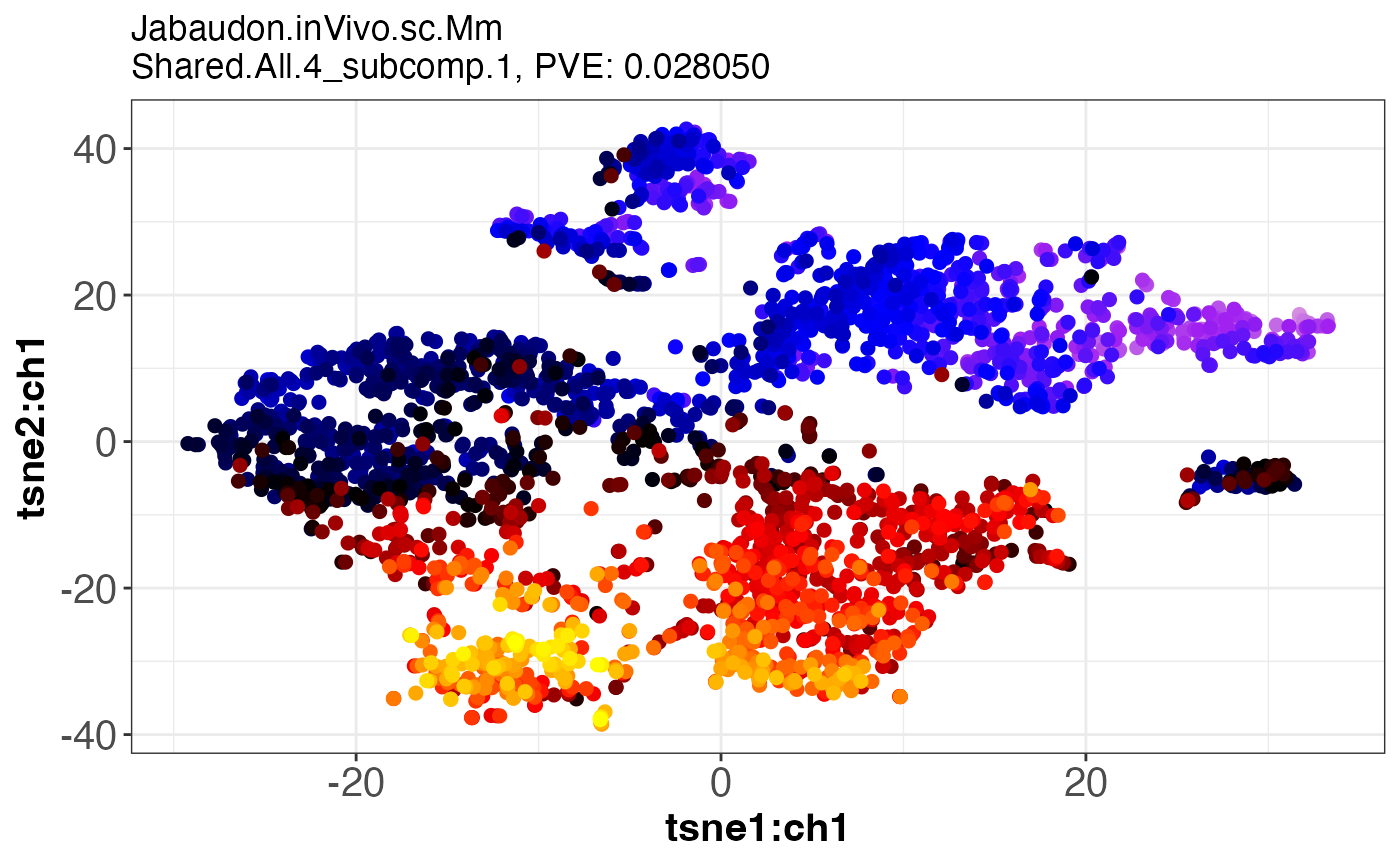
#> [1] "dataset: Jabaudon.inVivo.sc.Mm"
#> [1] "grouping: Shared.All.4"
#> [1] 1
#> [1] 2
#> [1] "grouping: Shared.bulk.2"
#> [1] "No SJD scores for grouping : Shared.bulk.2, moving to the next SJD output for Jabaudon.inVivo.sc.Mm"
#> [1] "grouping: Shared.sc.2"
#> [1] 1
#> [1] 2
#> [1] "grouping: Hs.Meisnr.1"
#> [1] "No SJD scores for grouping : Hs.Meisnr.1, moving to the next SJD output for Jabaudon.inVivo.sc.Mm"
#> [1] "grouping: Hs.AZ.1"
#> [1] "No SJD scores for grouping : Hs.AZ.1, moving to the next SJD output for Jabaudon.inVivo.sc.Mm"
#> [1] "grouping: Gesch.1"
#> [1] "No SJD scores for grouping : Gesch.1, moving to the next SJD output for Jabaudon.inVivo.sc.Mm"
#> [1] "grouping: Telley.1"
#> [1] 1
#> [1] 2
print(names(SJDScorePlotter.obj))
#> [1] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Meissner.inVitro.bulk.Hs.comp1of2"
#> [2] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Meissner.inVitro.bulk.Hs.comp2of2"
#> [3] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.bulk.2.data_Meissner.inVitro.bulk.Hs.comp1of2"
#> [4] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.bulk.2.data_Meissner.inVitro.bulk.Hs.comp2of2"
#> [5] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Hs.Meisnr.1.data_Meissner.inVitro.bulk.Hs.comp1of2"
#> [6] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Hs.Meisnr.1.data_Meissner.inVitro.bulk.Hs.comp2of2"
#> [7] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_LIBD.AZ.inVitro.bulk.Hs.comp1of2"
#> [8] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_LIBD.AZ.inVitro.bulk.Hs.comp2of2"
#> [9] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.bulk.2.data_LIBD.AZ.inVitro.bulk.Hs.comp1of2"
#> [10] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.bulk.2.data_LIBD.AZ.inVitro.bulk.Hs.comp2of2"
#> [11] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Hs.AZ.1.data_LIBD.AZ.inVitro.bulk.Hs.comp1of2"
#> [12] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Hs.AZ.1.data_LIBD.AZ.inVitro.bulk.Hs.comp2of2"
#> [13] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Geschwind.inVivo.sc.Hs.comp1of2"
#> [14] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Geschwind.inVivo.sc.Hs.comp2of2"
#> [15] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.sc.2.data_Geschwind.inVivo.sc.Hs.comp1of2"
#> [16] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.sc.2.data_Geschwind.inVivo.sc.Hs.comp2of2"
#> [17] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Gesch.1.data_Geschwind.inVivo.sc.Hs.comp1of2"
#> [18] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Gesch.1.data_Geschwind.inVivo.sc.Hs.comp2of2"
#> [19] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Jabaudon.inVivo.sc.Mm.comp1of2"
#> [20] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Jabaudon.inVivo.sc.Mm.comp2of2"
#> [21] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.sc.2.data_Jabaudon.inVivo.sc.Mm.comp1of2"
#> [22] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.sc.2.data_Jabaudon.inVivo.sc.Mm.comp2of2"
#> [23] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Telley.1.data_Jabaudon.inVivo.sc.Mm.comp1of2"
#> [24] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Telley.1.data_Jabaudon.inVivo.sc.Mm.comp2of2"Save the whole list to a .RData file
# save(SJDScorePlotter.obj, file = "./SJDScorePlotter.obj.RData")Assemble the images based on dataset
assemble.byDataset.obj = assemble.byDataset(SJDScorePlotter.obj = SJDScorePlotter.obj, dataset_name = "Meissner.inVitro.bulk.Hs", SJD_algorithm = "twoStageLCA", group = NA)
print(names(assemble.byDataset.obj))
#> [1] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Meissner.inVitro.bulk.Hs.comp1of2"
#> [2] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Meissner.inVitro.bulk.Hs.comp2of2"
#> [3] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.bulk.2.data_Meissner.inVitro.bulk.Hs.comp1of2"
#> [4] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.bulk.2.data_Meissner.inVitro.bulk.Hs.comp2of2"
#> [5] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Hs.Meisnr.1.data_Meissner.inVitro.bulk.Hs.comp1of2"
#> [6] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Hs.Meisnr.1.data_Meissner.inVitro.bulk.Hs.comp2of2"Plot all images corresponding from the
assemble.byDataset.obj. We can construct them into a whole
big image through the grid.arrange from the
grid.Extra package.
assemble.byDataset.obj[[1]]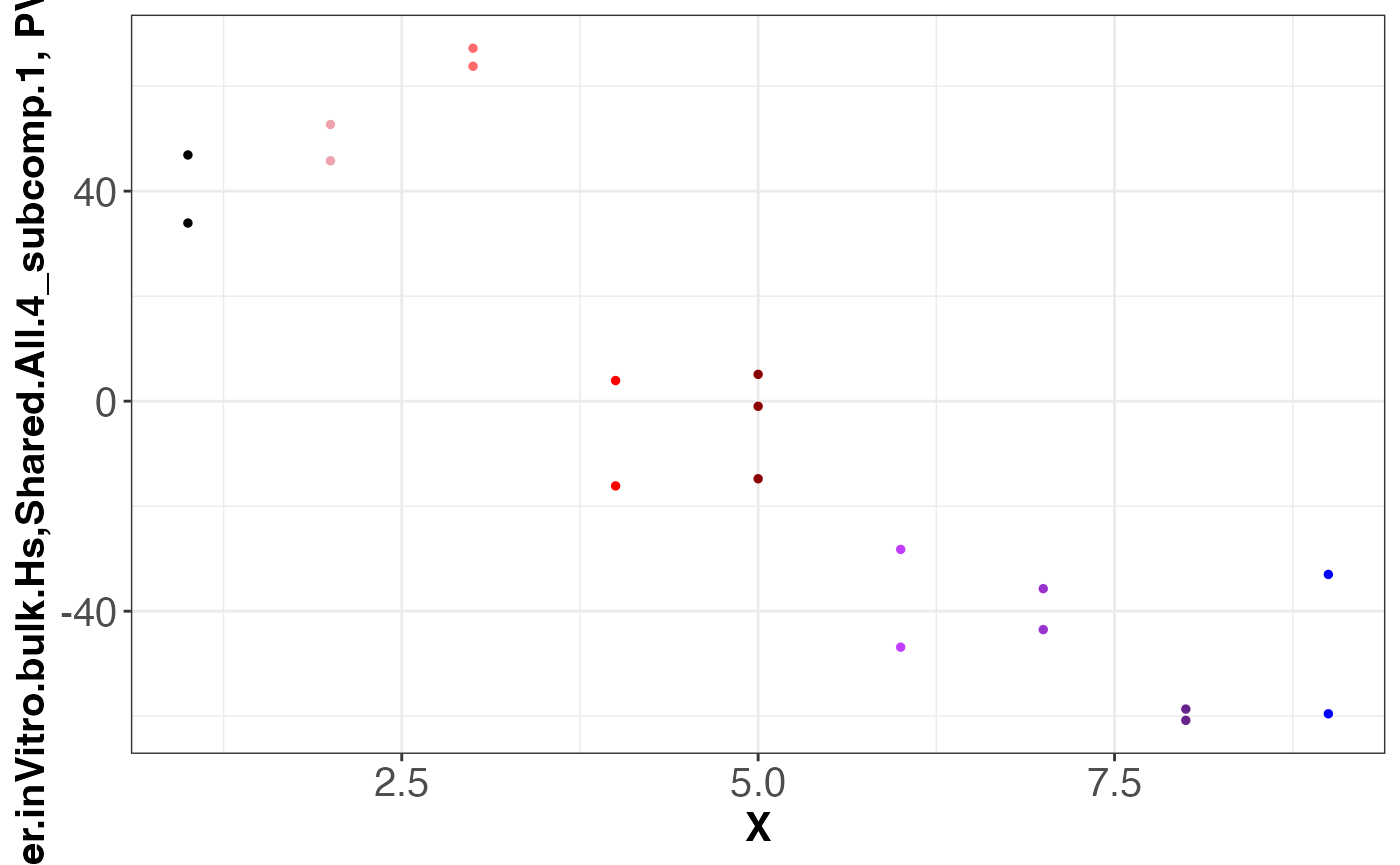
assemble.byDataset.obj[[2]]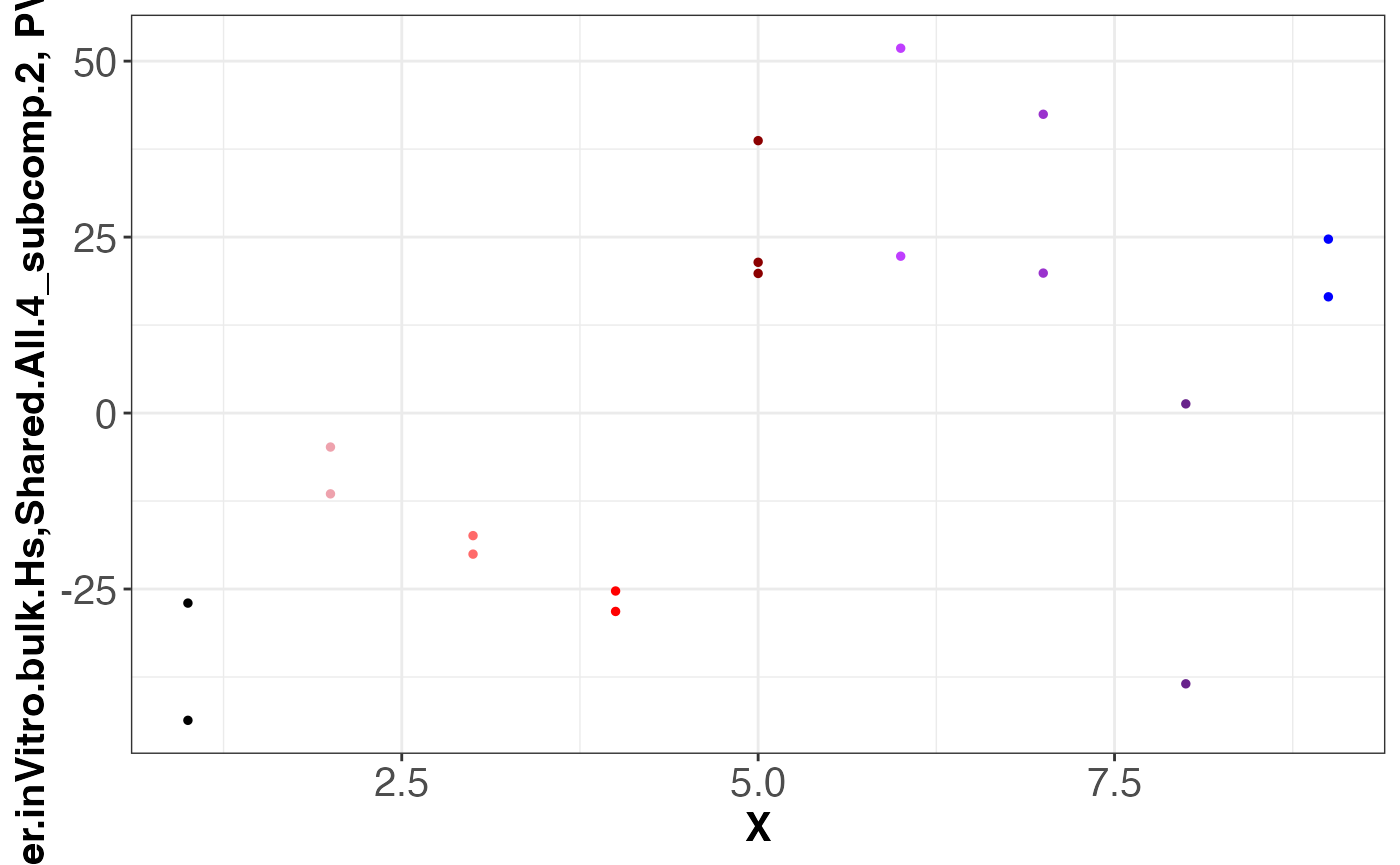
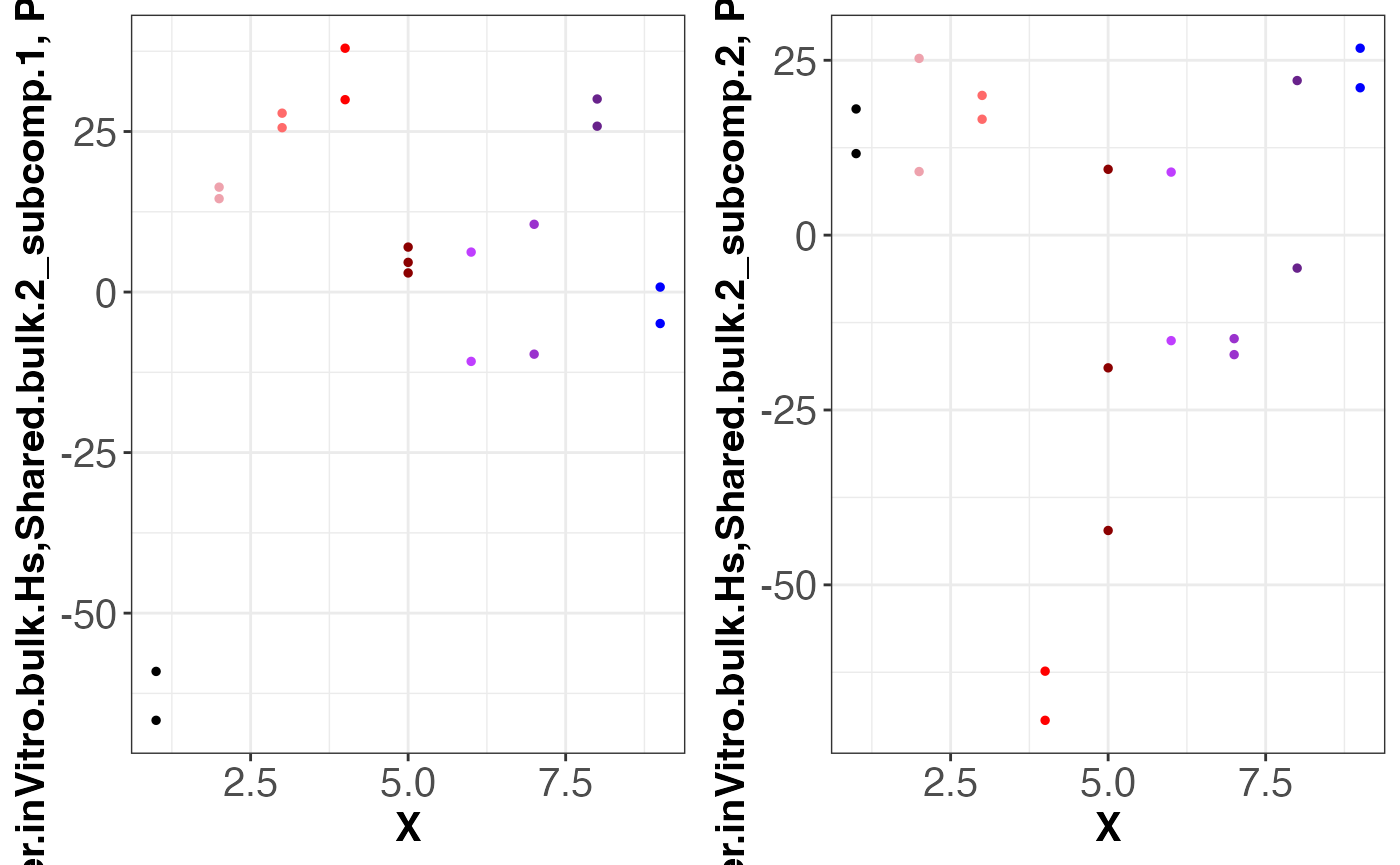
g = grid.arrange(
assemble.byDataset.obj[[3]],
assemble.byDataset.obj[[4]],
nrow = 1, ncol = 2
)
Assemble the images based on component
assemble.byComponent.obj = assemble.byComponent(SJDScorePlotter.obj = SJDScorePlotter.obj, component = c(1, 2), SJD_algorithm = "twoStageLCA", group = 'Shared.All.4')
print(names(assemble.byComponent.obj))
#> [1] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Meissner.inVitro.bulk.Hs.comp1of2"
#> [2] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_LIBD.AZ.inVitro.bulk.Hs.comp1of2"
#> [3] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Geschwind.inVivo.sc.Hs.comp1of2"
#> [4] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Jabaudon.inVivo.sc.Mm.comp1of2"
#> [5] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Meissner.inVitro.bulk.Hs.comp2of2"
#> [6] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_LIBD.AZ.inVitro.bulk.Hs.comp2of2"
#> [7] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Geschwind.inVivo.sc.Hs.comp2of2"
#> [8] "SJDout_NeuroGenesis4.p2.SJDalg_twoStageLCA.grp_Shared.All.4.data_Jabaudon.inVivo.sc.Mm.comp2of2"
assemble.byComponent.obj[[1]]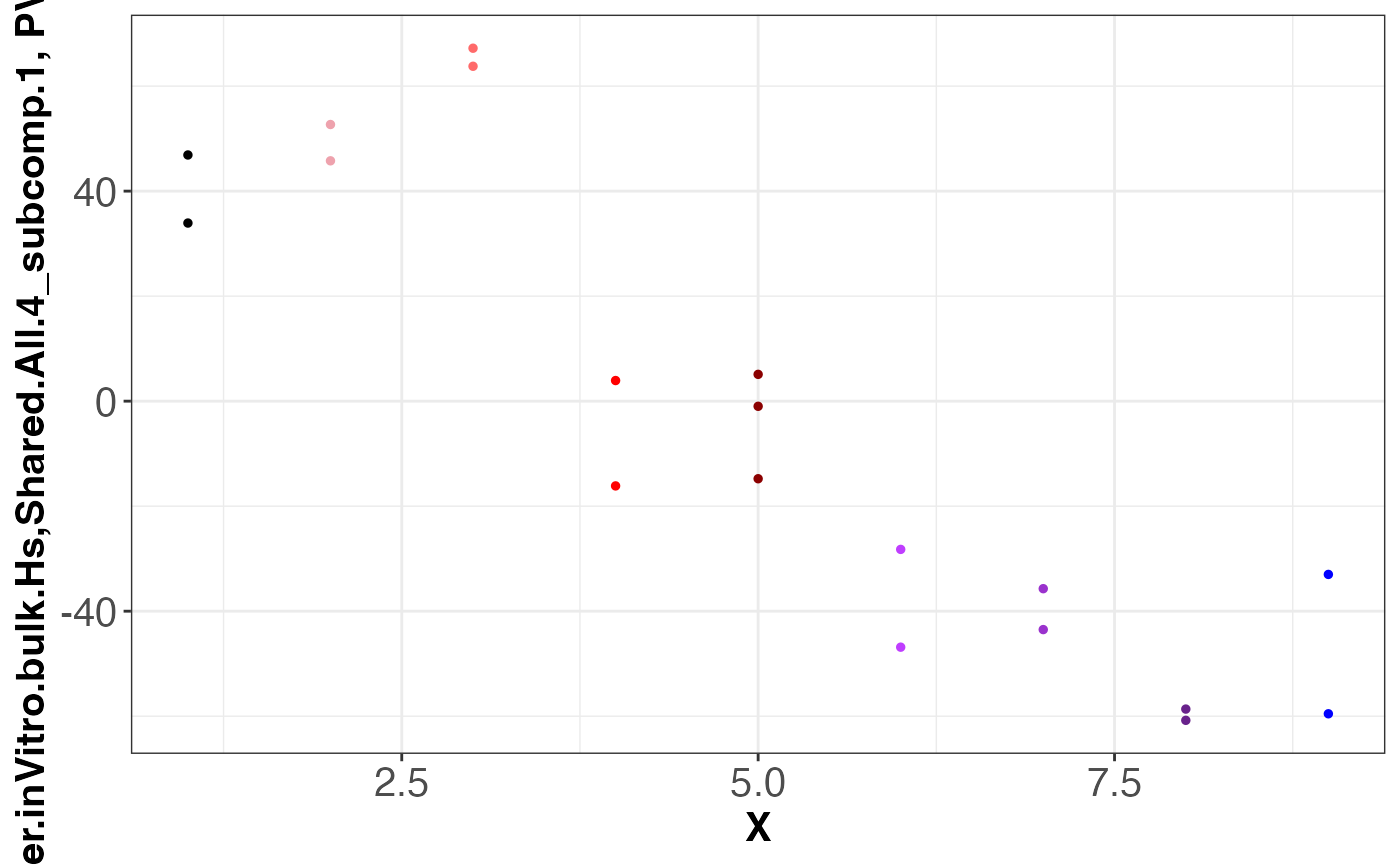
assemble.byComponent.obj[[2]]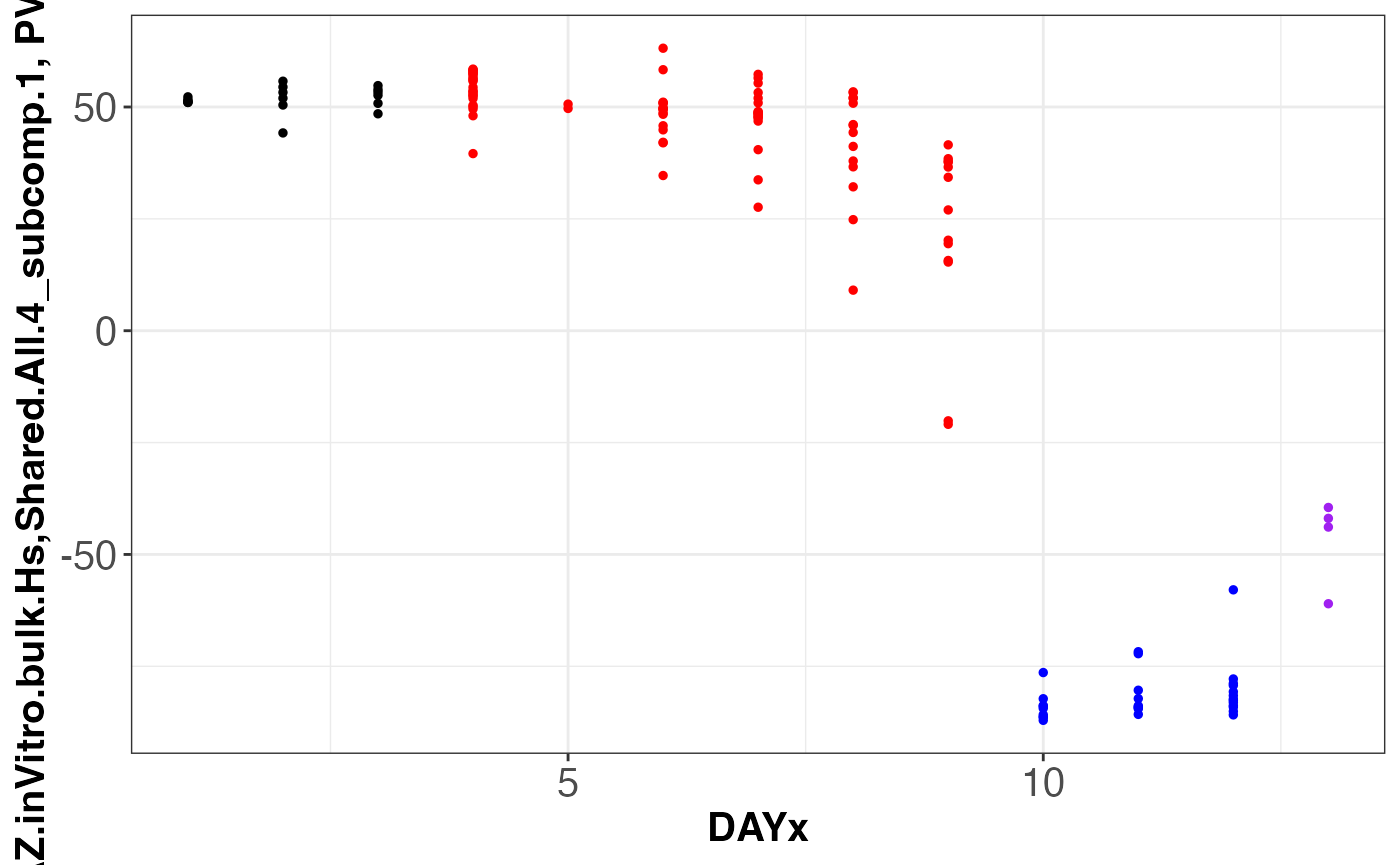
assemble.byComponent.obj[[3]]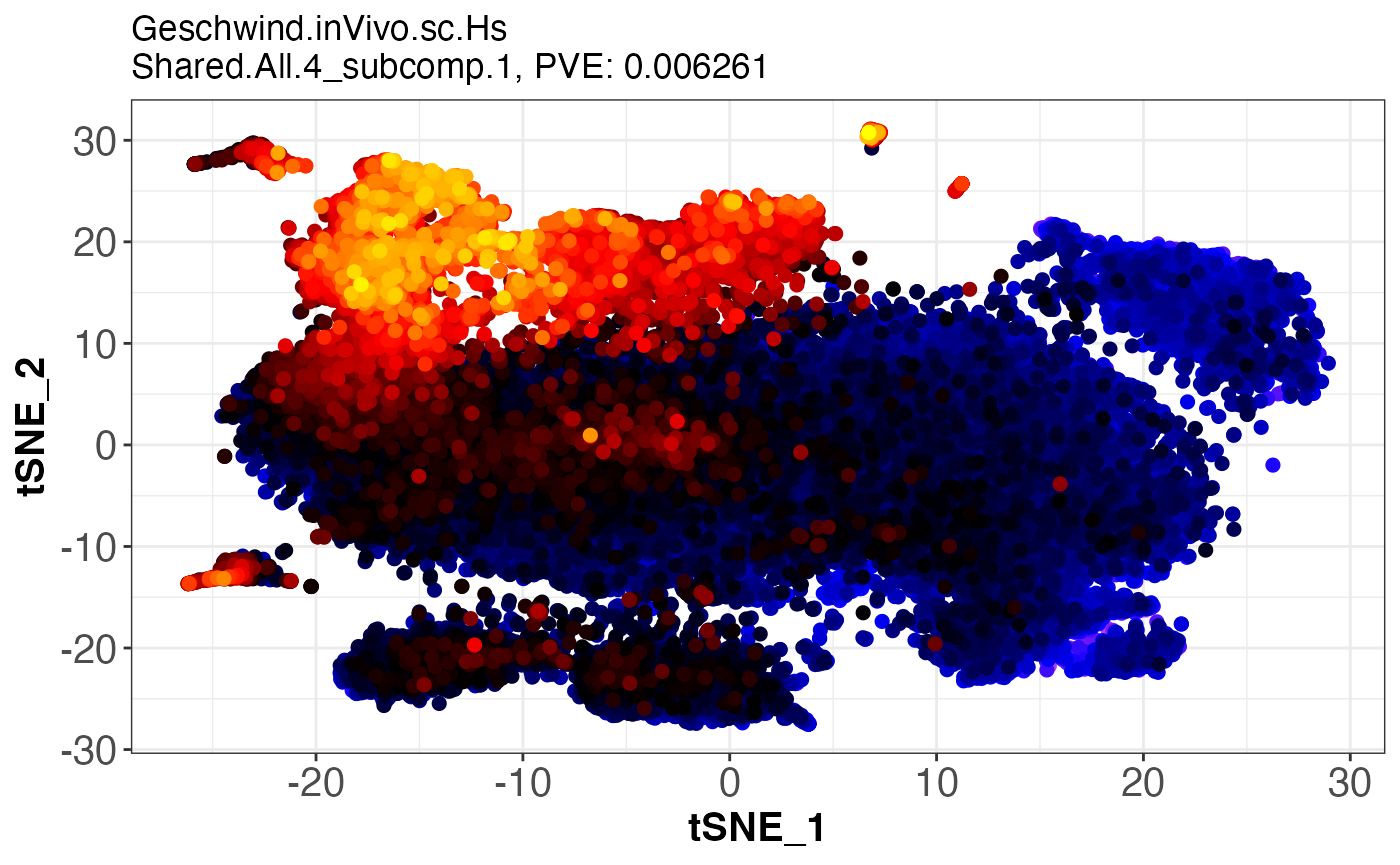
assemble.byComponent.obj[[4]]